
Abstract
Inflammasome activation is associated with disease severity in patients who are infected with SARS-CoV-2 and influenza viruses, but the specific cell types involved in inflammasome activation, as well as the balance of inflammasome activation versus viral replication in COVID-19 exacerbation and the induction of patient death, are unknown. In this study, we assessed lung autopsies of 47 COVID-19 and 12 influenza fatal cases and examined the inflammatory profiles and inflammasome activation; additionally, we correlated these factors with clinical and histopathological patient conditions. We observed an overall stronger inflammasome activation in lethal cases of SARS-CoV-2 compared to influenza and found a different profile of inflammasome-activating cells during these diseases. In COVID-19 patients, inflammasome activation is mostly mediated by macrophages and endothelial cells, whereas in influenza, type I and type II pneumocytes contribute more significantly. An analysis of gene expression allowed for the classification of COVID-19 patients into two different clusters. Cluster 1 (n=16 patients) died with higher viral loads and exhibited a reduced inflammatory profile than Cluster 2 (n=31 patients). Illness time, mechanical ventilation time, pulmonary fibrosis, respiratory functions, histopathological status, thrombosis, and inflammasome activation significantly differed between the two clusters. Our data demonstrated two distinct profiles in lethal cases of COVID-19, thus indicating that the balance of viral replication and inflammasome-mediated pulmonary inflammation may lead to different clinical conditions, yet both lead to patient death. An understanding of this process is critical for decisions between immune-mediated or antiviral-mediated therapies for the treatment of critical cases of COVID-19.
Introduction
Influenza viruses and epidemic coranaviruses are the most frequent viral agents of acute respiratory distress syndrome (ARDS). Between 2019 and 2021, there were 35,000,000 symptomatic cases of influenza with approximately 20,000 deaths in the United States 1 and more than 255,000,000 confirmed cases and 5,127,000 deaths caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) worldwide 2. Severe ARDS is characterized by major pulmonary involvement, respiratory distress, systemic thrombosis, and death 3–5.
The activation of the NLRP3 inflammasome has been described both in influenza 6–10 and SARS-CoV-2 infections 11–16, and this process contributes to excessive inflammation and poor clinical outcome. The recruitment of immune cells to the lungs of infected individuals culminates in the excessive release of cytokines that cause structural damage to the lungs 17, 18. Moreover, the major reported lung pathologies of severe COVID-19 include diffuse alveolar damage (DAD), acute fibrinoid organizing pneumonia (AFOP), and chronic interstitial pneumonia 3–5, 19. NLRP3 inflammasome activation also occurs in the lung parenchyma of these patients 16, 20. For influenza, the major reported lung pathologies include diffuse alveolar damage in addition to necrotizing bronchitis, bronchiolitis, and fibrosis 21. However, it is still unknown as to how the activation of this immune complex is related to the exacerbated inflammatory process in human lung parenchyma cells during viral infections such as influenza and SARS-CoV-2.
In the present study, postmortem lung tissue samples from 47 fatal COVID-19 patients and 12 fatal influenza patients were examined for inflammasome activation and gene expression; in addition, these factors were correlated with the clinical conditions of the patients to understand the molecular mechanisms underlying the pathological processes that lead to the death of these patients. We demonstrated the presence of robust inflammasome activation in the lungs of patients infected with SARS-CoV-2 and influenza virus and defined the specific cell types that contribute to inflammasome activation in response to these viruses. An analysis of gene expression in pulmonary tissues allowed for the classification of COVID-19 patients into two different clusters (one cluster with patients who died with higher viral loads and reduced inflammatory profiles, which opposes the data from the other cluster). Our data indicated that inflammasome activation by different cell types contributes to the pathology of influenza and COVID-19, thus indicating that a distinct balance in viral replication and inflammation may culminate with patient death in COVID-19.
Materials and Methods
Samples
Minimally invasive autopsy was performed on 47 patients diagnosed with SARS-CoV-2 at the Hospital das Clínicas da Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo, Brazil (Ribeirão Preto, SP, Brazil) from April to July, 2020, by Serviço de Patologia (SERPAT). Minimally invasive autopsy was done at bedside through post-mort surgical lung biopsy by a matching 14-gauge cutting needle (Magnum Needles, Bard) and a biopsy gun (Magnum, Bard). Moreover, a 3 cm incision on the more affected side of the chest between the fourth and fifth ribs were also used to provide extra lung tissue. All tissue samples were embedded in paraffin and fixed in formalin (Formalin-Fixed Paraffin-Embedded, FFPE). Lung tissue from autopsies of patients diagnosed with Influenza (n=12) of patients who died between the years 2012 to 2020 and biopsy of Lung Adenocarcinoma patients (n=5) were obtained from SERPAT. The project was approved by the Research Ethics Committee of FMRP/USP under protocol n° 4,089 .567.
Immunohistochemistry (IHC)
Tissue section in paraffin blocks were tested by immunohistochemistry using antibodies for the detection of NLRP3 (clone D2P5E; 1:3,000; Cell Signaling), ASC (1:2,000; Adipogen AL177), SFTPC (1:200, ThermoFisher PA5-71680), CD68 (1:200, Dako M0814), CD34 (1:500, Zeta Z2063ML), PDPN (1:200, ThermoFisher 14-9381-82), THY-1 (1:300, Sigma SAB4200497), Vimentin (1:200, Sigma V2258), Anti-cleaved N-terminal GSDMD (1:200, Abcam ab215203), Caspase-1 (1:200, Abcam ab207802), IL-1β (1:200, Abcam ab2105) for the in situ detection of these inflammasome proteins. The Sequential Immunoperoxidase Labeling and Erasing (SIMPLE) technique was used to evaluate all markers in the same tissue, as previously published 22. Briefly, after incubation with primary antibody (overnight at 4°C), slides were incubated with immune peroxidase polymer anti-mouse visualization system (SPD-125, Spring Bioscience, 345 Biogen) and with chromogen-substrate AEC peroxidase system kit (SK-4200, 346 Vector Laboratories, Burlingame, CA). After high resolution scanning by a VS120 Olympus microscope, the coverslips were removed in PBS and the slides were dehydrated in an ethanol gradient to 95% ethanol. The slides were incubated in a series of ethanol to erase the AEC marking. Afterwards the slides were rehydrated and the antibodies were removed with an incubation for 2 min in a solution of 0.15 mM 351 KMnO4/0.01 M H2SO4, followed immediately by a wash in distilled water. Tissues were then remarked.
Immunofluorescence
The slides were incubated with the primary antibodies, rabbit anti-human NLRP3 mAb (clone D2P5E; 1:300; Cell Signaling), rabbit anti-human ASC polyclonal antibody (1:200; Adipogen AL177), SFTPC (1:200, ThermoFisher PA5-71680), CD64 (1:200, Biolegend 139304), CD34 (1:500, Zeta Z2063ML), PDPN (1:200, ThermoFisher 14-9381-82), overnight at 4°C and with the secondary antibodies Goat anti-mouse Alexa fluor-647 (Invitrogen) or Goat anti-rabbit Alexa fluor-594 (Invitrogen). Images were acquired by the Axio Observer system combined with the LSM 780 confocal device microscope at 63x magnification (Carl Zeiss).
Histological Evaluation
Paraffin-embedded lung tissues sections (3 μm) were stained by standard hematoxylin and eosin (H&E). Morphological lung injury patterns were evaluated by specialized pulmonary pathologists (ATF) blinded to clinical history. They were classified as absent or present with its extent of lung injury area by 5% cut-offs in Fibrosis, Organizing Pneumonia (OP), Acute Fibrinous and Organizing Pneumonia (AFOP), Diffuse Alveolar Damage (DAD), Cellular Pneumonitis, Thrombus Formation.
RNA extraction
RNA from influenza samples and respective controls was extracted from Paraffin-embedded tissue samples. Samples were cut into 3 sections of 10 μm, and placed in RNase-free, 2.0-ml Eppendorf tubes. Sections were deparaffinized by incubation in 0.8 ml of xylene at 37°C for 5 minutes. The samples were then centrifuged, the supernatant was removed, and fresh xylene was added for a second incubation. After deparaffinization and centrifugation, sections were washed with 0.8 ml ethanol, air-dried for several minutes, and resuspended in 25 μl of 20 mg/ml Proteinase K (Gibco BRL, Gaithersburg, MD) plus 720 μl of a digestion buffer with the following final concentrations: 20mM TRIS-HCl pH8, 10mM EDTA pH8, 1% of SDS. Samples were vortexed and incubated overnight at 55°C at 400 rpm. A second 25 μl aliquot of 20 mg/ml Proteinase K was then added, followed by vortexing and incubation for 2h at 55°C. RNA was obtained by extraction with 1 mL of Trizol reagent and purification was performed according to the manufacturer’s instructions. Total RNA from fresh lung tissue of SARS-CoV-2 patients and respective controls was obtained using Trizol reagent and purification was performed according to the manufacturer’s instructions. The RNA was quantified by spectrophotometry in a NanoDrop 2000c spectrophotometer. The concentration was adjusted to 1 μg/µL, and the RNA was stored at −70 ◦C until reverse transcription.
Real-Time Polymerase Chain Reaction for inflammatory genes
For inflammatory genes qPCR the total RNA was transcribed into complementary DNA (cDNA) using a High-Capacity cDNA Reverse Transcription kit (without an inhibitor) according to the protocol provided by the manufacturer (Thermo Fisher, Carlsbad, CA, USA). The reaction was prepared in a final volume of 20.0 µL containing 4.2 µL of H2O, 2.0 µL of buffer, 2.0 µL of random primers, 0.8 µL of dNTP Mix (100 mM), 1.0 µL of reverse transcriptase (RT) enzyme and 1 µL of RNA (1 ug/µL). The solution was then placed into a thermocycler with the following program: 25 ◦C for 10 min, 37 ◦C for 120 min and 85 ◦C for 5 min. The real-time PCR was performed in 96-well plates using Sybr Green reagents (Applied Biosystems, Waltham, MA, USA) and a Quant studio real-time PCR system (Applied Biosystems, Foster City, CA, USA). The real time-RT-PCR was carried out in a total volume of 20 μl on a 96-well MicroAmp Fast Optical plate (Applied Biosystems). Each well contained 10 μl SYBR Green qPCR Master Mix (Thermofisher), 1 μl of each primer (Supplementary Table 1), 2 μl cDNA (20 ng) and 7 μl RNase free water using the following protocol: initial denaturation at 95°C for 10 min, 40 cycles of denaturation at 95°C for 15 s followed by annealing/extension at 60°C for 60 s. Each PCR was followed by a dissociation curve analysis between 60-95°C. The Ct values were analyzed by the comparative Ct (ΔΔCt) method and normalized to the endogenous control GAPDH. Fold difference was calculated as 2−ΔΔCt
Real-Time Polymerase Chain Reaction for Viral RNA
Detection and quantification of SARS-CoV-2 genes was performed with primer-probe sets for 2019-nCoV_N2 and gene E, according to US Centers for Disease Control and Prevention 23 and Charité group protocols 24. The genes evaluated (N2, E, and RNase-P housekeeping gene) were tested by one-step real-time RT-PCR using total nucleic acids extracted with TRIzol (Invitrogen). All real-time PCR assays were done on a Quant studio real-time PCR system (Applied Biosystems, Foster City, CA, USA). A total of 70 ng of RNA was used for genome amplification, adding specific primers (20 µM), and probe (5 µM), and with TaqPath 1-Step quantitative RT-PCR Master Mix (Applied Biosystems), with the following parameters: 25°C for 2 min, 50°C for 15 min, and 95°C for 2 min, followed by 45 cycles of 94°C for 5 s and 60°C for 30 s. Primers used were the following: N2 forward: 5′-TTACAAACATTGGCCGCAAA-3′, N2 reverse: 5′-GCGCGACATTCCGAAGAA-3′; N2 probe: 5′-FAM-ACAATTTGCCCCCAGCGCTTCAG-BHQ1-3′ 23; E forward: 5′-ACAGGTACGTTAATAGTTAATAGCGT-3′, E reverse: 5′-ATATTGCAGCAGTACGCACACA-3′; E probe: 5′-AM-ACACTAGCCATCCTTACTGCGCTTCG-BHQ-1-3′ 24; RNase-P forward: 5′-AGATTTGGACCTGCGAGCG-3′, RNase-P reverse: 5′-GAGCGGCTGTCTCCACAAGT-3′; and RNase-P probe: 5′-FAM-TTCTGACCTGAAGGCTCTGCGCG-BHQ-1-3′ 23. A plasmid of N2 protein and E protein was used for a standard curve construction for viral load quantification.
Chest computed tomography images acquisition and evaluation
Chest x-radiography (CXR) and computed tomography (CT) exams were performed as part of the routine clinical evaluation. Chest radiographies were performed in conventional equipment, mainly in the anteroposterior incidence. CT images were performed in multidetector scanners (Brilliance CT Big Bore 16 - Philips, Holland, or Aquilion Prime 160 - Toshiba, Japan), using similar protocols for the acquisition of high-resolution images of the lungs 25. Patients were scanned in the supine position without the administration of intravenous contrast media. Typical acquisition parameters were: 120 kVp tube voltage, 100–140 ref mAs 26, 0.3–0.7 s gantry rotation time, reconstruction matrix size of 512×512, slice thickness, and increment of 1.0 mm, using standard (soft) and hard kernel filters. Imaging exams were independently evaluated by two thoracic radiologists (MKS and DTW), blinded to clinical data, laboratory, and pathology results as described 26. Divergences were solved by consensus. All CXRs available were classified for the presence and grade of viral pneumonia 27. Pulmonary disease evolution on imaging was evaluated considering all exams from the initial to the last image before death. Tomographic images were evaluated similarly to CXR images.
Statistical Analysis
For puncta quantification, all histological sections were viewed on a 63x objective for digitalizing random images using the LSM 780 system in the Axio Observer microscope, covering an area of about ∼1.7 mm2 of lung parenchyma analyzed per case. Manual counting of puncta and cells was blinded and performed using the acquired images. Morphometric analyzes were performed as described 28. The quantification of expression by immunohistochemistry was performed by calculating the percentage of marked area, the scanned images were opened in the ImageJ software, using the IHQ Toolbox plugin, which consists of a semi-automatic color selection tool that selects the pixels positive for immunohistochemical marking, differentiating them from background and H&E marking, after selecting the positive pixels, the images were transformed into 8-bits and the area occupied by these tones was calculated. The distribution of the gene expression and biochemical marker and puncta count data was evaluated using the Shapiro–Wilk test; the data were analyzed using non-parametric Kruskal-Wallis, Mann-Whitney test and Spearman correlation. Data were presented in graphs with mean and standard deviation. Statistical analyzes were performed using the GraphPad PRISM 5.0 program, with p<0.05 being considered statistically significant. Heatmaps were constructed using the heatmap.2 function in the R program (Project for Statistical Computing, version 3.4.1).
Results
Lethal cases of COVID-19 develop severe acute respiratory distress syndrome (ARDS) and higher inflammasome activation in the lung parenchyma than influenza patients
We evaluated inflammasome activation in the lung parenchyma of 47 patients who died from SARS-CoV-2 infection from April to July 2020. These patients were infected with the ancestral strain of SARS-CoV-2 before the development of the COVID-19 variants of concern and before the use of vaccinations. We also evaluated 12 patients who died from influenza before the COVID-19 pandemic. As a noninfected control, we evaluated inflammasome activation in biopsies of the benign area of the lungs from patients who died due to lung adenocarcinoma. The data in Table 1 show the demographic and clinical characteristics of the influenza and COVID-19 patients. COVID-19 patients had a higher mean age (67.97 years) than influenza patients (41.5 years). Most of the influenza patients were infected with subtype A H1N1 (41.66%) but also with subtypes A H3N2 (16.66%) and B (16.66%). In addition, according to the data observed in the COVID-19 cases that occurred in 2020 (patients infected with the ancestral Wuhan strain of SARS-CoV-2), we found that COVID-19 patients had a higher frequency of comorbidities, such as hypertension, in addition to having (on average) grade 1 obesity (BMI: 31.10).
Influenza and COVID-19 patients characteristics
Furthermore, patients with influenza and COVID-19 (on average) exhibited altered values of CRP, D-dimers, LDH, creatinine, urea, AST, ALT, PT (INR), and blood glucose levels. Moreover, patients with influenza presented with a fulminant picture of ARDS and died (on average) less than 24 hours after seeking health services. In contrast, patients with COVID-19 had an average illness time of 18 days, and the vast majority (76.59%) of the patients used MV, with an average of 12.38 days of use of mechanical ventilation. Moreover, COVID-19 patients had a more severe respiratory condition than influenza patients, with a considerably altered PaO2 and PaO2/FiO2 characterizing a moderate to severe ARDS condition (Table 1).
Consistent with the respiratory statuses of the patients, the histopathological analyses of COVID-19 patients demonstrated a higher fibrotic phase of DAD, OP, and pneumonitis than those of influenza patients (Figure 1A-C). To analyze histological features of patients’ lungs, we measured the lung parenchyma area in the histological sections. In support of the data described above, we observed that patients who died from SARS-CoV-2 and influenza had a much larger area of lung parenchyma than adenocarcinoma patients, but no difference was found in influenza versus COVID-19, thus supporting the idea that both viral infections trigger robust inflammatory infiltration to the lung in severe cases of the disease (Figure 1D-G). These data are in agreement with histopathological findings showing an intense inflammatory process in patients who died from SARS-CoV-2 and influenza 3, 29.

Histopathological analysis of lethal cases of COVID-19 and Influenza patients. (A-G) Histological analysis of hematoxylin and eosin (H&E) stained slides for the presence of fibrosis (A), organizing pneumonia (B) and pneumonitis (C). (D-G) Proportion of lung parenchyma area of COVID-19, Influenza and adenocarcinoma (benign area of the lungs) patients. (E-G) Representative images of H&E stain showing the lung parenchyma. Scale bars 200 µm. (H-K) Multiphoton microscopy analysis of lung autopsies of 47 COVID-19 patients, 12 influenza patients and 5 lung adenocarcinoma patients. Tissues were stained with anti-ASC (H, I) or anti-NLRP3 (J, K) for quantification of cells with inflammasome puncta in lung autopsies (in red, indicated by white arrows). DAPI stains cell nuclei (blue). Insets indicate a higher magnification of the indicated region (red rectangle). Scale bars 20 µm. (L) Percentage of NLRP3 and ASC puncta colocalization in the lungs of five COVID-19 patients. (M) Representative images showing ASC (green) and NLRP3 (red) colocalization. Scale bar 10 µm. Each dot in the figure represents the value obtained from each individual. *, P < 0.05 comparing the indicated groups, as determined by Mann-Whitney test (A-D) or by Kruskal-Wallis test (H, J). The images were acquired by multiphoton microscope using a 63x oil immersion objective and analyzed using ImageJ Software
To assess inflammasome activation in the lungs of COVID-19 and influenza patients, the lung parenchyma area and cell count were scanned in histological sections by using 2-photon microscopy. The parenchyma area was calculated by using ImageJ software, and inflammasome activation was microscopically scored by the presence of characteristic NLRP3 or ASC puncta/specks 30. The parenchyma area was used to normalize all of the counts between the patients. Inflammasome activation was quantified by counting ASC and NLRP3 puncta in the lung tissue of these patients. We observed that patients with SARS-CoV-2 infection had more ASC and NLRP3 puncta than patients with influenza and controls with lung adenocarcinoma (Figure 1H, J). Representative images of ASC and NLRP3 puncta in the patients’ lungs are shown (Figures 1I, K). We observed ASC colocalization in nearly all of the scored NLRP3 puncta, thus confirming that these puncta structures that were abundantly found in the patients’ lungs are indeed the NLRP3/ASC inflammasome (Figure 1L, M). Collectively, these data indicate that COVID-19 patients have higher inflammasome activation than influenza patients and that these patients evolved to a fibrotic phase of DAD, OP, and pneumonitis.
The cellular profile of inflammasome activation differs in lethal cases of COVID-19 and influenza
To comprehensively investigate the cell types operating during influenza and SARS-CoV-2 infection, we analyzed the presence of macrophages (CD64+), endothelial cells (CD34+), type 1 pneumocytes (PDPN+, Podoplanin+), and type 2 pneumocytes (SFTPC+) in the lungs of patients who died from SARS-CoV-2 infection and influenza. We found that total counts of macrophages and endothelial cells were not different between COVID-19 and influenza patients and were also similar to those observed in the benign area of lung adenocarcinoma (Figure 2A, 2B). However, we observed that patients who died from influenza had fewer type 1 pneumocytes and type 2 pneumocytes (Figure 2C, 2D), which is consistent with data indicating that the influenza virus preferentially infects type 1 and type 2 pneumocytes than macrophages and promotes the death of these cells due to the viral replication process 31. Next, we evaluated the participation of specific cells in inflammasome activation by scoring ASC and NLRP3 puncta in macrophages (CD64+), endothelial cells (CD34+), type 1 pneumocytes (PDPN+), and type 2 pneumocytes (SFTPC+).

Multiphoton microscopy analysis of lung autopsies of 47 COVID-19 patients, 12 influenza patients and 5 lung adenocarcinoma patients (benign area of the lungs). (A-D) Numbers of macrophages (CD64+, A), endothelial cells (CD34+, B), type I pneumocytes (PDPN+, C) and type II pneumocytes (SFTPC+, D) per lung parenchyma area. (E-L) Percentage CD64+ (E, I), CD34+ (F, J), PDPN+ (G, K) and SFTPC+ (H, L) containing ASC (E-H) or NLRP3 (I-L) puncta. Each dot in the figures represents the value obtained from each individual. *, P < 0.05 comparing the indicated groups, as determined by Kruskal-Wallis test. (M-T) Representative image of ASC (M-P) or NLRP3 (Q-T) puncta (in red; indicated by white arrows) in CD64+ (in red; M, Q), CD34+ (in purple; N, R), PDPN+ (in green; O, S) and SFTPC+ (in green; P, T) cells from a lung autopsy of a COVID-19 patient. DAPI stains cell nuclei (blue). Scale bars 10 µm. The images were acquired by multiphoton microscopy using a 63x oil immersion objective and analyzed using ImageJ Software.
We observed that in the lungs of COVID-19 patients, both ASC and NLRP3 puncta were more frequently found in macrophages (Figure 2E, I) and endothelial cells (Figure 2F, J) than in influenza patients. In contrast, in the assessment of type 1 and type 2 pneumocytes, we found that ASC and NLRP3 puncta were more frequently observed in the lungs of influenza patients than in COVID-19 patients (Figure 2G, H, K, L). Representative images of ASC and NLRP3 puncta in macrophages, endothelial cells, and type 1 and type 2 pneumocytes are shown in Figure 2M-T. Together, these data demonstrate for the first time that distinct pulmonary cell types can trigger inflammasome activation in COVID-19 and influenza; additionally, the data indicate a main contribution of macrophages and endothelial cells to inflammasome activation during SARS-CoV-2 and type I and II pneumocytes to inflammasome activation during influenza. The magnitude of inflammasome activation and the different cellular profiles of inflammasome activation between COVID-19 and influenza may contribute to explaining the overall increased inflammasome activation found in COVID-19 compared to influenza.
Subsequently, we performed immunohistochemistry analyses of the lung parenchyma to assess the expression of inflammasome components and observed variable expression of NLRP3, ASC, cleaved GSDMD, IL-1β, and caspase-1 in COVID-19 and influenza (Supplementary Fig. 1A-E). To assess which lung parenchyma cells expressed these inflammasome components, we evaluated the expression of ASC, NLRP3, caspase-1, cleaved GSDMD, and IL-1β on endothelial cells (CD34+), macrophages (CD68+), and type 2 pneumocytes (SFTPC+) via immunohistochemistry (Supplementary Fig. 1F-H). We also stained these tissues with anti-Spike of SARS-CoV-2 and found that virus-associated endothelial cells, macrophages, and type 2 pneumocytes expressed ASC, NLRP3, Caspase-1, and IL-1β (Supplementary Fig. 1F-H).
Disease progression in lethal cases of COVID-19 occurs with decreasing viral load and increasing inflammasome activation over time in the lung parenchyma
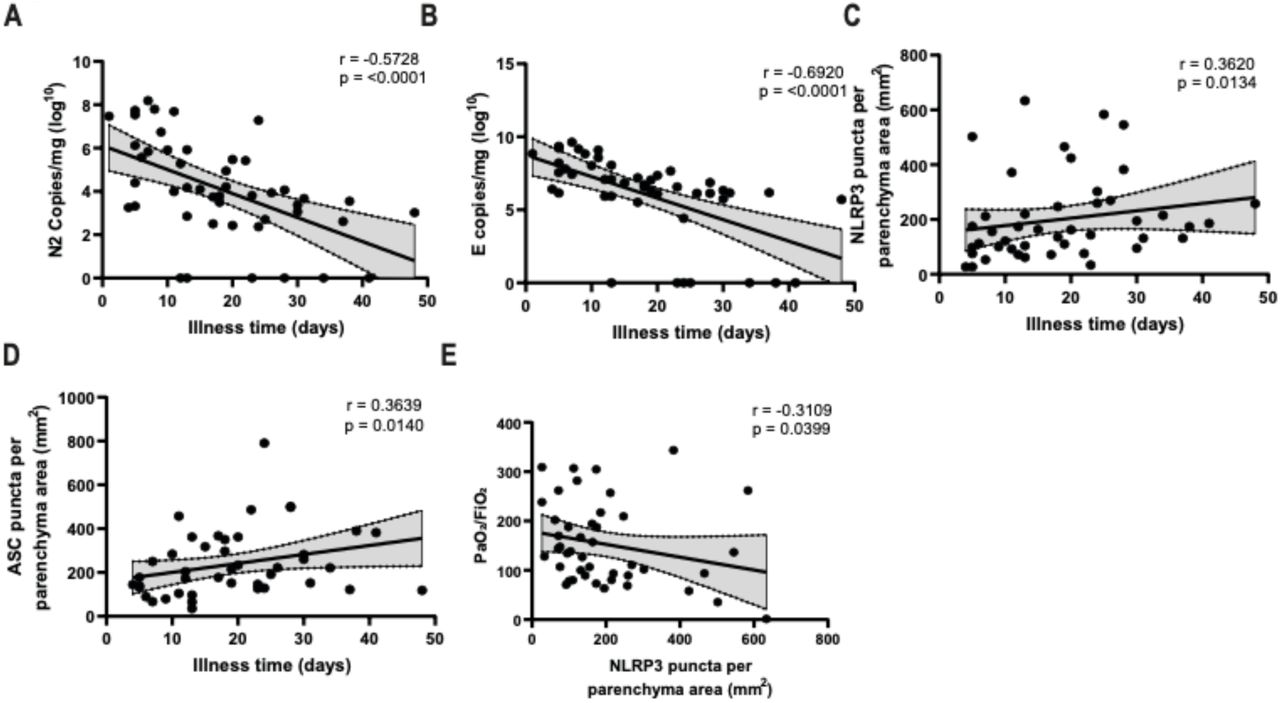
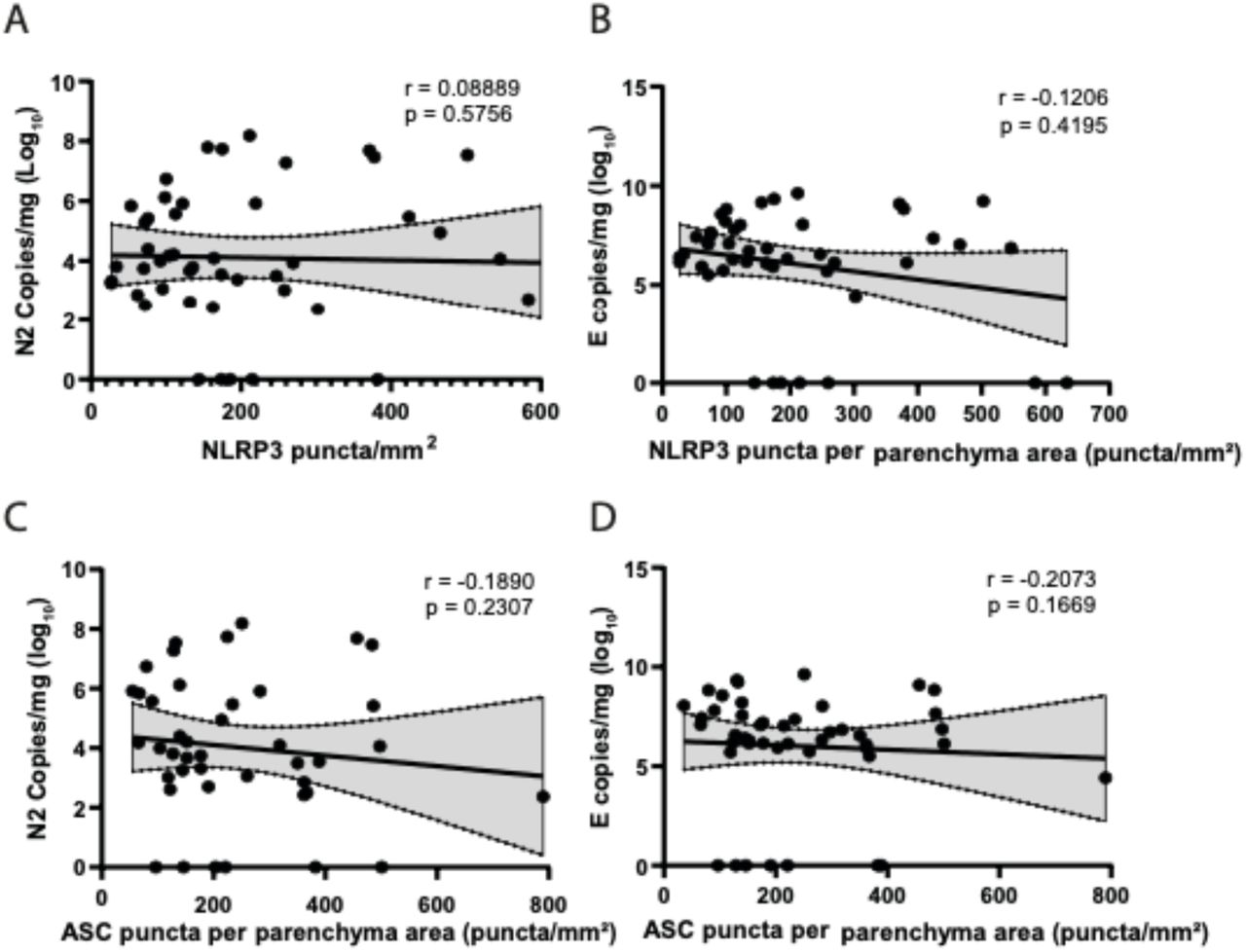
To assess whether inflammasome activation was associated with specific patient clinical conditions, we performed Pearson’s correlations in COVID-19 patients. Even though all of these patients died, we observed a strong negative correlation between viral load and the extent of the disease (Figure 3A, B). In addition, we observed a positive correlation between the amount of NLRP3 and ASC puncta with the time of disease (Figure 3C, D), thus suggesting that, in general, whereas the viral load is reduced, inflammasome activation increases during hospitalization in these lethal cases of COVID-19. We also observed significant negative correlations between NLRP3 puncta and PaO2/FiO2 (Figure 3E), thus suggesting that inflammasome activation is related to an overall worsening pulmonary function. We detected no statistically significant correlations between NLRP3 or ASC puncta and viral load in the tissues (Supplementary Fig. 2).

Spearman correlation of pulmonary viral load, inflammasome activation and illness time in 47 fatal COVID-19 patients. (A) Correlation of viral N2 with illness time; (B) Correlation of viral E with illness time; (C) Correlation of NLRP3 puncta per parenchyma area with illness time; (D) Correlation of ASC puncta per parenchyma area with illness time; (E) Correlation of NLRP3 puncta per parenchyma area with PaO2/FiO2. r and p-value are indicated in the figure.
Inflammasome activation and pulmonary viral loads defines two distinct clinical outcomes in lethal cases of COVID-19
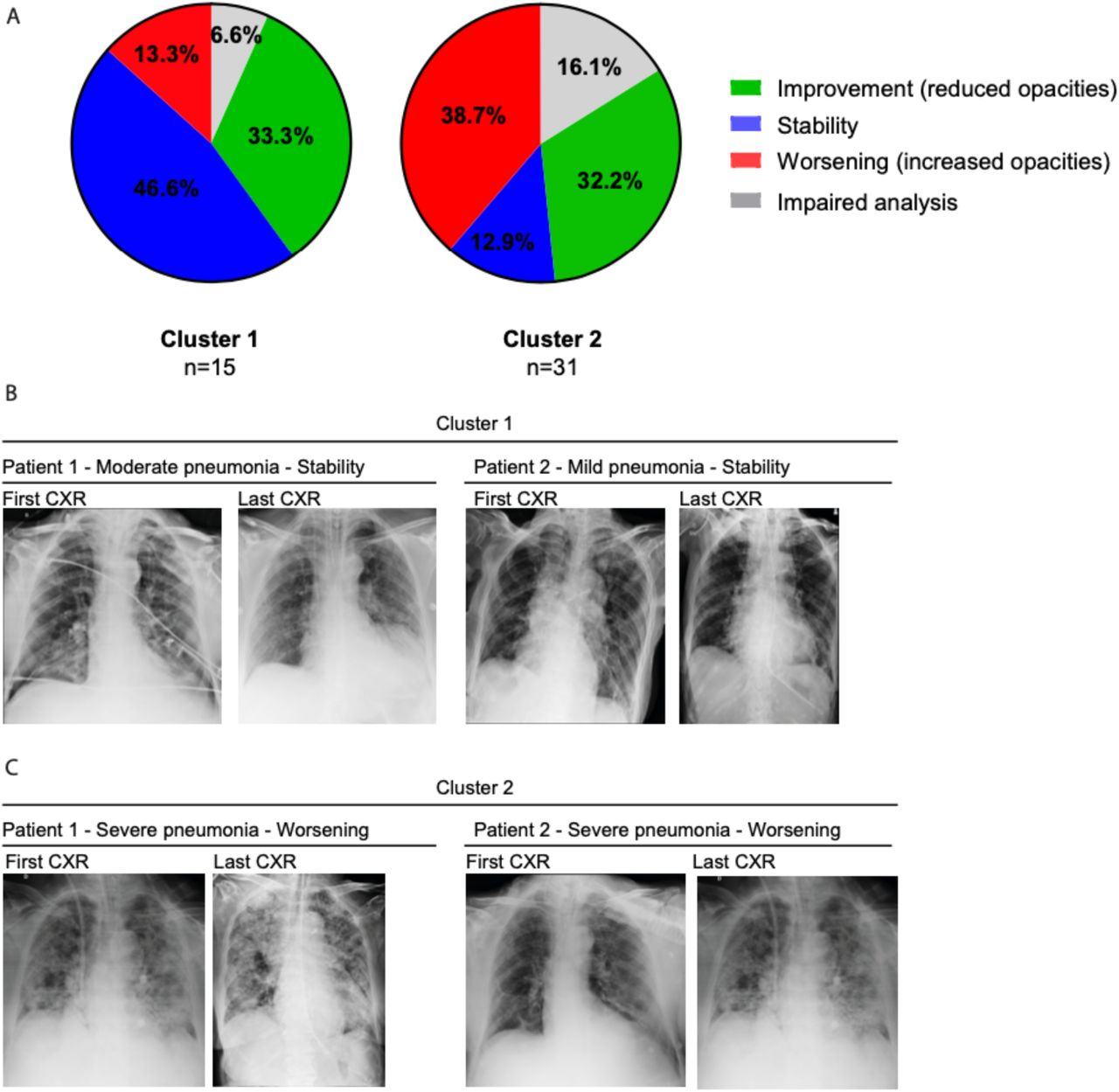
The imbalance of inflammatory and anti-inflammatory processes leads to an excessive release of cytokines into the systemic circulation with potentially deleterious consequences, including systemic inflammatory response syndrome (SIRS), circulatory shock, multiorgan dysfunction syndrome (MODS), and death 32. Several studies have reported the association of influenza 33–37 and SARS-CoV-2 38–40 with hyperinflammatory syndrome related to disease severity, and inflammasome activation may be associated with Cytokine Release Syndrome (CRS) 41–43. Thus, to correlate inflammasome activation with the hyperinflammatory profile observed in lethal cases of COVID-19, we analyzed the expression of genes related to the inflammatory process and inflammasome activation. We evaluated gene expression in the lungs of COVID-19 patients and observed that several inflammatory cytokine genes have a positive correlation with the genes involved in inflammasome activation (Supplementary Fig. 3A). We did not observe a significant difference in the expression of these genes when we compared COVID-19 patients with lung adenocarcinoma patients (Supplementary Fig. 3B-U), possibly due to the large dispersion of data observed in COVID-19 patients. Due to this dispersion and high variation in gene expression detected in COVID-19 patients, we performed an unsupervised heatmap constructed with data from relative gene expression and revealed the formation of two clusters in COVID-19 patients (Figure 4A). Cluster 1 was characterized by a higher viral load and lower expression of inflammasome and inflammatory genes. In contrast to Cluster 1, Cluster 2 was comprised of patients who died with a lower viral load and increased expression of inflammasome and inflammatory genes (Figure 4A). To gain insights into key differences between these two clusters, we analyzed PaO2/FiO2 and A-a O2 gradient kinetics in these patients. We observed that patients in Cluster 2, who had overall increased inflammation, had worsening pulmonary function compared to those in Cluster 1 (Figure 4B, C). By contrast, patients of Cluster 1 had disseminated intravascular coagulation (Table 2), according to the parameters recommended by the International Society of Thrombosis and Hemostasis (ISTH) 44. Basic demographic information stratified by cluster is provided in Table 2. Strikingly, patients who belonged to Cluster 2 had a longer illness time (Figure 4D), worse pulmonary function (as indicated by the PaO2/FiO2 and A-a O2 gradients) (Figure 4E, F), a larger area of pulmonary fibrosis (Figure 4G), and greater activation of the NLRP3 inflammasome, as measured by puncta formation (Figure 4H). Representative images of the lung parenchyma of three representative patients in Cluster 1 and three representative patients in Cluster 2 are shown in Figure 4I-J. In addition, we confirmed that patients belonging to Cluster 2 had an overall lower viral load than patients from Cluster 1 (Supplementary Fig. 4A, B). Next, we assessed radiological analyses of 16 patients from Cluster 1 and 31 patients from Cluster 2 by comparing the first and last chest X-ray images (CXR) and observed overall worsening pulmonary conditions in patients from Cluster 2 (Supplementary Fig. 5A). Representative images of CXR of 2 patients from each cluster are shown in Supplementary Fig. 5B, C. Table 3 summarizes the general imaging evaluation findings for the patients in Cluster 1 and Cluster 2. Taken together, our data suggest the existence of two distinct groups of patients who succumbed to COVID-19, one group, with lower viral loads, higher inflammation, increased fibrosis and worse pulmonary conditions than the other group.

(A) Heatmap of the mRNA expression of inflammasome, inflammatory molecules/cytokines and viral N2 and E in lung autopsies of 47 COVID-19 patients. PaO2/FiO2 (B) and A-a O2 gradient (C) during disease development of patients from Cluster 1 (N=16, green) and Cluster 2 (N=31, red). *, P < 0.05 comparing the indicated groups, as determined by Area Under Curve test. (D-H) Analysis of Cluster 1 and Cluster 2 for illness time (D), PaO2/FiO2 (E), A-a O2 gradient (F), fibrosis (G), and NLRP3 puncta per lung parenchyma area (H). Each dot in the figure represents the value obtained from each individual. *, P < 0.05 comparing the indicated groups, as determined by Mann-Whitney test. (I-J) Representative H&E images of lung parenchyma of 3 patients of cluster 1 (I) and 3 patients of cluster 2 (J). Scale bars 200 µm.
COVID-19 (Cluster 1 and Cluster 2) patients characteristics
Discussion
Although SARS-CoV-2 and influenza share many similarities, including the transmission, replication, and induction of similar clinical outcomes in humans, COVID-19 can be significantly more severe in nonvaccinated individuals 45–48. The mechanisms underlying these differences are unknown, and the revelation that exacerbated inflammasome activation mediates COVID-19 pathology 13, 14, 16 has led to comparisons of inflammasome activation in COVID-19 patients with other pulmonary viral diseases. Therefore, we assessed pulmonary samples of lethal cases of COVID-19 and influenza and determined the cellular profile of inflammasome activation in patients’ lungs. Lethal cases of COVID-19 exhibited an overall greater inflammasome activation, in addition to having a different cellular activation profile, compared to lethal cases of influenza. This different profile of inflammasome activation can be partially explained by the tropism of each virus. Whereas the influenza virus almost exclusively infects pneumocytes, with minor involvement of macrophages, SARS-CoV-2 associates with a broad range of immune cells, including macrophages and structural cells, such as type 2 pneumocytes, club-like cells, goblet cells, and endothelial cells 31, 49–52. Thus, it is possible that the large number of cell types that can be infected by SARS-CoV-2 may be a determining factor for the increased activation of the inflammasome observed in fatal cases of COVID-19 compared to influenza viruses. In addition, reduced inflammasome activation in influenza patients can be partially explained by the NS1 protein of the influenza A virus, which inhibits the NLRP3 inflammasome by suppressing ASC ubiquitination 53. Moreover, whereas a protective role of the inflammasome has never been reported for COVID-19, these platforms account for protective antiviral immunity against influenza infection 54, 55. Therefore, it is possible that influenza patients with higher inflammasome activation undergo a resolution of infection; as a result, these patients do not seek hospitalizations, as often occurs with COVID-19 patients.
Macrophages are possibly the most effective cell types that trigger inflammasome activation 56; according to this information, our data showed that macrophages in the lungs of COVID-19 and influenza patients are effectively infected and strongly induce inflammasome activation. These data are in agreement with previously published articles indicating high levels of viral RNA in lung monocytes and macrophages of COVID-19 patients 57, 58, as well as pronounced inflammasome activation in macrophages from COVID-19 patients 13, 14, 16. In contrast, the influenza virus, which effectively replicates in type I and type II pneumocytes, promotes exacerbated inflammasome activation and the depletion of these cells in the lungs of fatal cases of influenza. It is possible that the inflammasome and pyroptosis of type I and II pneumocytes during influenza infection contribute to the clinical symptoms and clinical outcomes of this disease. Importantly, this appears to be the first report of inflammasome activation in type 1 and type 2 pneumocytes in the lung, as the description of inflammasome activation in pneumocytes was previously restricted to the A549 cell lineage 59–61. Remarkably, inflammasome activation in pneumocytes is more pronounced in patients infected with the influenza virus. It is possible that inflammasome activation in pneumocytes is favored by influenza virus tropism in these cells 31, 49.
In addition to inflammasome activation in pneumocytes, the analysis of inflammasome activation in different pulmonary cell types indicates that endothelial cells exposed to SARS-CoV-2 express inflammasome proteins and contain active NLRP3/ASC puncta, thus demonstrating inflammasome activation. Despite reports indicating that endothelial cells are not productively infected by SARS-CoV-2 62, precursors of hematopoietic and endothelial cells stimulated with SARS-CoV-2 spike protein increase the expression of AIM2, NLRP1, NLRP3, IL1Β, and ASC and trigger caspase-1 activation 63, 64. In addition, inflammasome activation in endothelial cells has been previously reported 65–68. These observations are consistent with our data indicating the expression of inflammasome components and inflammasome activation in CD34+ cells in the lungs of COVID-19 patients. Although these cells were stained positive for the SARS-CoV-2 spike protein, it is unknown whether infection and viral replication are required for inflammasome activation in these cells. It is possible that spike stimulation is sufficient for inflammasome activation, which is a feature that would explain reports indicating damage and dysfunction of these cells during COVID-19 and the characterization of COVID-19 as an endothelial disease 69–74. It is important to highlight the fact that the vascular endothelium is actively involved in the regulation of inflammation and thrombus formation. This is particularly important in COVID-19 because the interplay between inflammasome activation in macrophages and the endothelium can lead to pyroptotic macrophages releasing tissue factor (TF), which is an essential initiator of coagulation cascades that are frequently observed in severe cases of COVID-19 75–77. These data support the association of inflammasomes with the coagulopathy that is observed in COVID-19.
Regardless of the cell type involved in inflammasome activation and comparisons with influenza, our robust analysis of 47 COVID-19 patients allowed for the classification of the patients into two groups. Cluster 2, which was composed of 31 patients, demonstrated remarkably high inflammasome activation and expression of inflammatory genes with increased fibrosis in patients’ lungs. Inflammatory cytokines released upon inflammasome activation have been previously linked to the development of pulmonary fibrosis 78–83. In addition, cytokines such as IL-1β, IL-18, and IL-1α have been described as triggering the activation of fibroblasts and stimulating the synthesis and accumulation of type I collagen, TIMP, collagenase, and PGE2 78, 84, which may contribute to the triggering of the deleterious effects of inflammasomes in patients’ lungs.
Our findings showed that the viral load in the lungs of COVID-19 patients is inversely proportional to the duration of the disease, as has been suggested in previous studies 85, 86. In addition, we collected data indicating that increased illness time is associated with increased inflammasome activation, and a reduced viral load implies a dysregulated immune response in which inflammation persists, whereas the viral load declines. In this context, it is possible that inflammasome activation in these patients occurs not as a result of the viral infection itself but by the presence of damage-associated molecular patterns that are generated during the inflammatory process. We also tested whether the mechanical ventilation used in COVID-19 patients would interfere with inflammasome activation. This hypothesis is supported by data indicating that mechanical ventilation-induced hyperoxia can induce potassium efflux through the P2X7 receptor, thus leading to inflammasome activation and the secretion of proinflammatory cytokines 43, 87–91. However, when we separated the COVID-19 patients into two groups according to the use or non-use of mechanical ventilation, we detected no differences in histopathological analyses and inflammasome activation (Table 4). Moreover, we confirmed that patients who underwent mechanical ventilation had a longer illness time, higher hypertension, less CRP and albumin, higher amounts of urea, and worsened pulmonary function (according to the PaO2/FiO2 and A-an O2 gradients). Thus, our data do not support the hypothesis that mechanical ventilation is directly associated with inflammasome activation. Nonetheless, we demonstrated (for the first time) a cellular profile of inflammasome activation in the lungs of patients who died from COVID-19 and influenza. Importantly, the identification of two distinct profiles in lethal cases of COVID-19 (which revealed the balance of viral loads versus inflammasome-mediated pulmonary inflammation) will be critical for decisions between immune-mediated or antiviral-mediated therapies for the treatment of critical cases of COVID-19.
Characteristics of covid-19 patients who were submitted or not to mechanical ventilation (MV).
Data Availability
All data produced in the present study are available upon reasonable request to the authors
Funding
Fundação de Amparo à Pesquisa do Estado de Sao Paulo, FAPESP grants 2013/08216-2, 2019/11342-6 and 2020/04964-8. Conselho Nacional de Desenvolvimento Científico e Tecnológico, CNPq grant 303021/2020-9. Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, CAPES grant 88887.507253/2020-00.
Competing interests
Authors declare that they have no competing interests.
Supplementary Figures and Supplementary Figure Legends

Immunohistochemistry analysis of lung autopsies of 47 COVID-19 patients, 12 influenza patients and 5 lung adenocarcinoma patients (benign area of the lungs). Quantification of percentage of area stained for ASC (A), NLRP3 (B), cleaved GSDMD (C), IL-1β (D) and Caspase-1 (E). (F-H) Representative images of lungs from fatal cases of COVID-19 and Influenza, indicating colocalization of viral Spike, ASC, NLRP3, Caspase-1, IL-1β and cleaved GSDMD in endothelial cells (CD34+, F), macrophages (CD68+, G), and type II pneumocytes (SFTPC+, H).

Spearman correlation of pulmonary viral load and inflammasome activation in 47 fatal COVID-19 patients. (A) Correlation of viral N2 with NLRP3 puncta per parenchyma area; (B) Correlation of viral E with NLRP3 puncta per parenchyma area; (C) Correlation of viral N2 with ASC puncta per parenchyma area; (D) Correlation of viral E with ASC puncta per parenchyma area. r and p-value are indicated in the figure.

matrix of inflammasome and inflammatory gene expression in lung autopsy of 47 COVID-19 patients (A). Colors indicate correlation scores, categorized as positive strong correlation (r ≥ 0.70; red); moderate positive correlation (0.50 ≥ r ≤ 0.70; orange); weak positive correlation (0.30 ≥ r ≤ 0.50; yellow); negative strong correlation (r ≥ −0.70; dark blue); negative moderate correlation (−0.50 ≥ r ≤ −0.70; blue) or negative weak correlation (−0.30 ≥ r ≤ −0.50; light blue). Only correlations with p<0.05 are represented in the correlation matrix. (B-U) Expression of mRNA in the lung autopsies of COVID-19 patients and lung adenocarcinoma patients (benign area of the lungs). Selected genes were Il6 (B), Il10 (C), Il17 (D), Ifna1 (E), Ifnb1 (F), Ifng (G), Il4 (H), Il2 (I), Tnfa (J), Il1a (K), Il1b (L), Il18 (M), I1ra (N), Nlrp3 (O), Nlrc4 (P), Nlrp1 (Q), Aim2 (R), Pycard (S), Casp1 (T), Casp4 (U).

Quantification of viral N2 (A) and E (B) in lung autopsies of 47 COVID-19 patients from Cluster 1 (N=16) and Cluster 2 (N=31). (C) Quantification of percentage of area stained for viral Spike protein in lung autopsies. Each dot in the figure represents the value obtained from each individual. *, P < 0.05 comparing the indicated groups, as determined by Mann-Whitney test. (D-E) Representative images of lungs tissues stained for Spike (red) and Hematoxylin (blue). Scale bars 100 µm

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Analysis of the first and last Chest x-radiography (CXR) of 46 COVID-19 patients belonging from Cluster 1 (n=15) and Cluster 2 (n=31). (A) patients with reduced opacities (green), stability (blue) and increased opacities (red) comparing the fists and first CXR. Impaired analyses are shown in gray. Representative images of first and last CXR from two patients from Cluster 1, indicating stability in moderate and mild pneumonia (B) and two patients from Cluster 2, indicating worsening conditions in cases of severe pneumonia (C).
Acknowledgments
We would like to thank Maira Nakamura, Amanda Zuin and Dr. Roberta Sales for technical support.
References
- 1.↵
- 2.↵
- 3.↵
- 4.
- 5.↵
- 6.↵
- 7.
- 8.
- 9.
- 10.↵
- 11.↵
- 12.
- 13.↵
- 14.↵
- 15.
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.
- 35.
- 36.
- 37.↵
- 38.↵
- 39.
- 40.↵
- 41.↵
- 42.
- 43.↵
- 44.↵
- 45.↵
- 46.
- 47.
- 48.↵
- 49.↵
- 50.
- 51.
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.
- 67.
- 68.↵
- 69.↵
- 70.
- 71.
- 72.
- 73.
- 74.↵
- 75.↵
- 76.
- 77.↵
- 78.↵
- 79.
- 80.
- 81.
- 82.
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.
- 89.
- 90.
- 91.↵



